Skip to content
Publications
2023
Extraction, labeling, clustering, and semantic mapping of segments from clinical notes
Petr ZELINA, Jana HALÁMKOVÁ, Vít NOVÁČEK
IEEE Transactions on Nanobioscience
This work is motivated by the scarcity of tools for accurate, unsupervised information extraction from unstructured clinical notes in computationally underrepresented languages, such as Czech. We introduce a stepping stone to a broad array of downstream tasks such as summarisation or integration of individual patient records, extraction of structured information for national cancer registry reporting or building of semi-structured semantic patient representations that can be used for computing patient embeddings. More specifically, we present a method for unsupervised extraction of semantically-labeled textual segments from clinical notes and test it out on a dataset of Czech breast cancer patients, provided by Masaryk Memorial Cancer Institute (the largest Czech hospital specialising exclusively in oncology). Our goal was to extract, classify (i.e. label) and cluster segments of the free-text notes that correspond to specific clinical features (e.g., family background, comorbidities or toxicities). Finally, we propose a tool for computer-assisted semantic mapping of segment types to pre-defined ontologies and validate it on a downstream task of category-specific patient similarity. The presented results demonstrate the practical relevance of the proposed approach for building more sophisticated extraction and analytical pipelines deployed on Czech clinical notes.
TLDR: We extend our work on finding structure in unstructured clinical notes with better segment grouping, analysis and ways to map segment types to existing dictionaries.
Unsupervised extraction, classification and visualization of clinical note segments using the MIMIC-III dataset
Petr ZELINA, Vít NOVÁČEK, Jana HALÁMKOVÁ
IEEE BIBM 2023
This paper presents a text-mining approach to extracting and organizing segments from unstructured clinical notes in an unsupervised way. Our work is motivated by the real challenge of poor semantic integration between clinical notes produced by different doctors, departments, or hospitals. This can lead to clinicians overlooking important information, especially for patients with long and varied medical histories. This work extends a previous approach developed for Czech breast cancer patients and validates it on the publicly accessible MIMIC-III English dataset, demonstrating its universal and language-independent applicability. Our work is a stepping stone to a broad array of downstream tasks, such as summarizing or integrating patient records, extracting structured information, or computing patient embeddings. Additionally, the paper presents a clustering analysis of the latent space of note segment types, using hierarchical clustering and an interactive treemap visualization. The presented results demonstrate that this approach generalizes well for MIMIC and English.
TLDR: We test, whether our clinical note segmentation approach works on an English dataset (MIMIC-III)
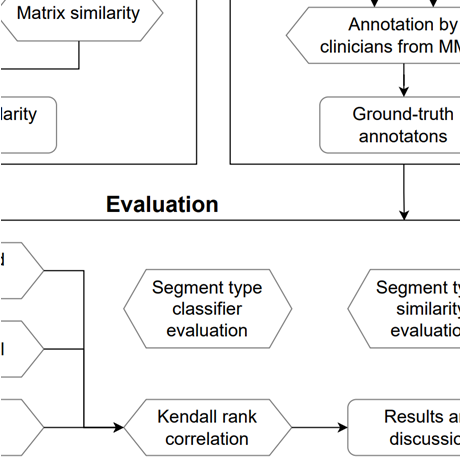
Patient similarity based on unstructured clinical notes
Petr ZELINA
Master's thesis
This thesis introduces a new way of measuring patient similarity based only on unstructured clinical notes. The system is able to focus on different aspects of patient similarity using a novel semi-supervised note-filtering technique. This approach has been tested on a dataset of Czech clinical notes of 4267 breast cancer patients. A validation study in cooperation with clinicians from the Masaryk Memorial Cancer Institute has been conducted and evaluated. The results show that this system is able to capture some patient similarity categories well, but more research needs to be done to predict others.
TLDR: I explore how to use unstructured clinical notes for calculating how similar are different pairs of patients.

Empowering Cancer Patients through Personalized Information: User-Centered Design of an E-Health System
Marko Řeháček
Master's thesis
Despite the growth in the number of breast cancer patients, there is still a lack of research focused on delivering digital solutions to cater their information needs. Patients often lack sufficient information about the disease and tend to feel disempowered. This thesis explores the design and development of a patient-centered eHealth application, which aims to empower breast cancer patients by providing them with comprehensible information about their condition, treatment, medication, and psychocognitive aspects of the care. The research was conducted in close collaboration with a clinical oncologist and the patients. Based on this collaboration, an application was designed and a prototype of the application was tested with the patients.
Machine learning-assisted recurrence prediction for patients with early-stage non-small-cell lung cancer
Adrianna Janik, Maria Torrente, Luca Costabello, Virginia Calvo, Brian Walsh, Carlos Camps, Sameh K Mohamed, Ana L Ortega, Vít Nováček, Bartomeu Massuti, and others
JCO Clinical Cancer Informatics
Stratifying patients with cancer according to risk of relapse can personalize their care. In this work, we provide an answer to the following research question: How to use machine learning to estimate probability of relapse in patients with early-stage non–small-cell lung cancer (NSCLC)? For predicting relapse in 1,387 patients with early-stage (I-II) NSCLC from the Spanish Lung Cancer Group data (average age 65.7 years, female 24.8%, male 75.2%), we train tabular and graph machine learning models. We generate automatic explanations for the predictions of such models. For models trained on tabular data, we adopt SHapley Additive exPlanations local explanations to gauge how each patient feature contributes to the predicted outcome. We explain graph machine learning predictions with an example-based method that highlights influential past patients. Machine learning models trained on tabular data exhibit a 76% accuracy for the random forest model at predicting relapse evaluated with a 10-fold cross-validation. Graph machine learning reaches 68% accuracy over a held-out test set of 200 patients, calibrated on a held-out set of 100 patients. Our results show that machine learning models trained on tabular and graph data can enable objective, personalized, and reproducible prediction of relapse and, therefore, disease outcome in patients with early-stage NSCLC. With further prospective and multisite validation, and additional radiological and molecular data, this prognostic model could potentially serve as a predictive decision support tool for deciding the use of adjuvant treatments in early-stage lung cancer.
TLDR: We used machine learning to predict the risk of cancer recurrence in patients with early-stage non-small cell lung cancer. By training models on patient data (both in tables and as graphs), we found that these AI methods can accurately estimate the probability of relapse, which can help doctors personalize treatment and improve patient outcomes.
2022
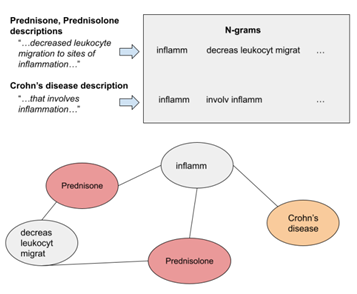
Text-Enhanced Relational Learning for Drug Repurposing
Marek TOMA
Master's thesis
Drug repurposing is a strategy for the development of new treatment options with already existing drugs. Computational approaches based on machine learning can further reduce the cost and accelerate the drug development process. In this work, we develop relational learning models for the prediction of associations between drugs and diseases. First, we design a model trained on the biomedical knowledge graph. Then, we develop two multimodal extensions, that incorporate textual data into the model. Lastly, we compare the performance of our models and analyze the quality of their predictions, showing promising results for some diseases, and we discuss the limitations and their possible solutions.
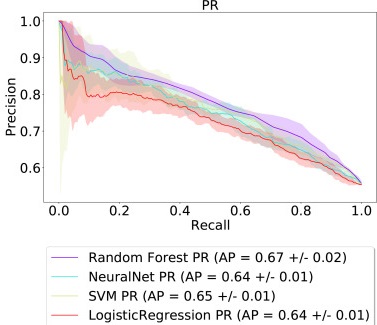
On predicting recurrence in early stage non-small cell lung cancer
Sameh K Mohamed, Brian Walsh, Mohan Timilsina, Maria Torrente, Fabio Franco, Mariano Provencio, Adrianna Janik, Luca Costabello, Pasquale Minervini, Pontus Stenetorp, and others
AMIA Annual Symposium Proceedings
Early detection and mitigation of disease recurrence in non-small cell lung cancer (NSCLC) patients is a non-trivial problem. This preliminary study describes an experimental suite of various machine learning models applied to a patient cohort of 2442 early-stage NSCLC patients to predict recurrence. We provide the outcomes of our assessment of basic supervised machine learning methods, evaluating these models on a binary classification task to classify patients with successful treatments into one of two categories. The results show that the random forest model achieved the best results in terms of all the used evaluation metrics. We discuss the promising results achieved, as well as the lessons learned while developing this baseline for further, more advanced studies in this area. Our results show that machine learning models trained on tabular and graph data can enable objective, personalized, and reproducible prediction of relapse and, therefore, disease outcome in patients with early-stage NSCLC. With further prospective and multisite validation, and additional radiological and molecular data, this prognostic model could potentially serve as a predictive decision support tool for deciding the use of adjuvant treatments in early-stage lung cancer.
TLDR: We used various machine learning models to predict if non-small cell lung cancer will return in patients after early-stage treatment. Our study, using data from over 2400 patients, shows that these models, especially random forest, can effectively predict recurrence, offering a valuable tool for personalized treatment decisions and improving patient outcomes.
An artificial intelligence-based tool for data analysis and prognosis in cancer patients: results from the clarify study
Maria Torrente, Pedro A Sousa, Roberto Hernácdez, Mariola Blanco, Virginia Calvo, Ana Collazo, Gracinda R Guerreiro, Beatriz Nunez, Joao Pimentao, Juan Cristobal Sánchez, and others
Cancers
Simple prognostic biomarkers are critical in clinical practice but often overlook the multi-parametric nature of cancer. This study explores the potential of an **Artificial Intelligence (AI)-based tool** to improve data analysis and prognosis in cancer patients, presenting results from the **CLARIFY study**. The CLARIFY study is a real-world, multicenter, observational study collecting extensive clinical, pathological, and molecular data from various cancer types. The AI tool integrates and analyzes this complex, heterogeneous data to identify patterns and generate more accurate prognostic predictions than traditional methods. Specifically, the tool leverages machine learning algorithms to uncover subtle relationships within the high-dimensional data, providing personalized risk stratification and insights into disease progression. Our findings demonstrate that this AI-driven approach significantly enhances prognostic accuracy, potentially leading to more informed treatment decisions and improved patient outcomes by considering the intricate interplay of multiple factors in cancer development and progression.
TLDR: We developed an AI tool to better analyze patient data and predict outcomes in cancer. Based on the CLARIFY study, this tool combines various types of patient information to provide more accurate and personalized prognoses, helping doctors make better treatment decisions.
2021
Biological applications of knowledge graph embedding models
Sameh K Mohamed, Aayah Nounu, Vít Nováček
Briefings in Bioinformatics
The rapid growth of biological data, combined with its inherent complexity and heterogeneity, necessitates advanced computational approaches for extracting meaningful insights. **Knowledge graph embedding (KGE) models** have emerged as powerful tools for representing and reasoning over vast biological knowledge. This review provides a comprehensive overview of the diverse **biological applications of knowledge graph embedding models**. We categorize these applications into key areas such as drug discovery and repurposing (e.g., drug-target interaction prediction, adverse drug reaction prediction), disease genomics (e.g., gene-disease association prediction, disease mechanism elucidation), protein-protein interaction prediction, and biological pathway analysis. We discuss the underlying principles of various KGE models, their advantages in handling complex biological relationships, and the challenges associated with their implementation in biological domains. Furthermore, we highlight recent advancements and future directions, emphasizing the transformative potential of KGEs in accelerating biological discovery and precision medicine.
TLDR: We review how advanced computer models called 'knowledge graph embeddings' are being used to solve big problems in biology. These models help make sense of vast amounts of biological data to, for example, find new drugs, understand diseases better, or predict how proteins interact, ultimately speeding up scientific discovery.
2020
Pretraining and Evaluation of Czech ALBERT Language Model
Petr ZELINA
Undergrad thesis
This thesis explores a new language model called ALBERT, released by Google Research in 2019. The ALBERT architecture has been very successful in English NLP tasks, and this thesis tests its applicability for the Czech language. The work gives an overview of the models leading to ALBERT and their design. Further, the creation process of several Czech ALBERT models is described. Finally, their performance is evaluated, and the models are compared with the current Czech state-of-the-art solutions for text classification and question answering tasks.
TLDR: Experiments with pre-training of a Czech variant of the ALBERT encoder-only transformer.
Discovering protein drug targets using knowledge graph embeddings
Sameh K Mohamed, Vít Nováček, Aayah Nounu
Bioinformatics
Motivation: Identifying protein targets for new or existing drugs is a crucial step in drug discovery and repurposing. Traditional experimental methods are time-consuming and expensive. Computational approaches, particularly those leveraging the growing volume of biomedical data, offer a promising alternative. Results: We propose a novel computational framework that utilizes knowledge graph embeddings (KGEs) for predicting drug–target interactions (DTIs). We construct a comprehensive knowledge graph by integrating diverse biomedical datasets, including information about drugs, proteins, diseases, and pathways. This graph serves as the foundation for learning low-dimensional vector representations (embeddings) of entities and relations using advanced KGE models. By treating DTI prediction as a link prediction task within this knowledge graph, our model can effectively identify novel interactions. We evaluated our method on a benchmark dataset and demonstrated superior performance compared to state-of-the-art computational methods, achieving high accuracy in predicting both known and potential novel drug-protein associations. This framework provides a powerful tool for accelerating drug discovery by efficiently prioritizing candidate drug targets for experimental validation.
TLDR: We developed a new computer method to find which proteins drugs might interact with (drug targets). We do this by building a large network of biomedical facts (a 'knowledge graph') and then using advanced AI to learn patterns within it. Our method can accurately predict new drug-protein interactions, which can help speed up the discovery of new medicines.
Accurate prediction of kinase-substrate networks using knowledge graphs
Vít Nováček, Gavin McGauran, David Matallanas, Adriáč Vallejo Blanco, Piero Conca, Emir Muñoz, Luca Costabello, Kamalesh Kanakaraj, Zeeshan Nawaz, Brian Walsh, and others
PLoS Computational Biology
Kinases are a crucial class of enzymes involved in almost all cellular processes, making the accurate prediction of kinase-substrate interactions (KSIs) vital for understanding cellular signaling and drug discovery. Traditional methods for identifying KSIs are often time-consuming and labor-intensive. In this study, we present a novel computational approach that leverages **knowledge graphs** to accurately predict kinase-substrate networks. We construct a comprehensive knowledge graph by integrating diverse biological data, including information about kinases, substrates, phosphorylation sites, and relevant biological pathways. By representing these entities and their relationships within a graph, we can apply **knowledge graph embedding (KGE)** techniques to learn latent representations that capture complex biological associations. We then use these embeddings to predict novel KSIs. Our extensive evaluation demonstrates that this knowledge graph-based approach significantly outperforms existing methods in predicting kinase-substrate interactions, providing a powerful tool for accelerating the discovery of new therapeutic targets and advancing our understanding of cellular regulation.
TLDR: We developed a new computer method to accurately predict how enzymes called kinases interact with other molecules (substrates). We did this by building a large network of biological information (a 'knowledge graph') and using advanced AI to find hidden patterns. Our method is much better at predicting these interactions, which is important for understanding how cells work and finding new drugs.
Predicting polypharmacy side-effects using knowledge graph embeddings
Vít Nováček, Sameh K Mohamed
AMIA Summits on Translational Science Proceedings
The increasing prevalence of polypharmacy (the concurrent use of multiple medications) in patient care leads to a growing concern about potential adverse drug-drug interactions, often manifesting as polypharmacy side-effects (PSEs). Predicting these complex interactions is a major challenge in clinical practice. This paper presents a novel computational approach to predict polypharmacy side-effects by leveraging **knowledge graph embeddings (KGEs)**. We construct a comprehensive knowledge graph that integrates information about drugs, diseases, symptoms, and known adverse drug reactions. By applying KGE models, we learn low-dimensional vector representations of entities and relationships within this graph. These embeddings are then used to predict novel and previously unobserved PSEs, treating the problem as a link prediction task. Our experiments demonstrate that this knowledge graph-based approach can effectively identify potential polypharmacy side-effects, offering a valuable tool for clinicians to enhance patient safety and optimize medication regimens. The method can help prioritize drug combinations for further investigation and reduce the risk of adverse events.
TLDR: We developed a computer system to predict harmful side effects when patients take multiple medications at once. By building a network of drug and health information ('knowledge graph') and using advanced AI, our system can identify potential problems, helping doctors prescribe safer drug combinations.
BioKG: A knowledge graph for relational learning on biological data
Brian Walsh, Sameh K Mohamed, Vít Nováček
ACM CIKM
The exponential growth of biological data across various modalities presents both opportunities and challenges for knowledge discovery. Integrating and making sense of this heterogeneous data is crucial for advancing biomedical research. In this paper, we introduce **BioKG**, a novel and comprehensive **knowledge graph specifically designed for relational learning on biological data**. BioKG integrates a wide array of biological entities and relationships from diverse public databases, including information on genes, proteins, diseases, drugs, pathways, and molecular interactions. We detail the schema and construction methodology of BioKG, emphasizing its rich semantic content and interconnectedness. Furthermore, we demonstrate the utility of BioKG by applying state-of-the-art knowledge graph embedding (KGE) models to perform various downstream tasks, such as drug-target interaction prediction and disease gene prioritization. Our results show that BioKG, combined with relational learning techniques, provides a powerful resource for inferring novel biological associations and accelerating discoveries in life sciences.
TLDR: We created **BioKG**, a new, large network of biological information (a 'knowledge graph') that connects different types of data like genes, drugs, and diseases. This graph is designed to help computers learn complex relationships within biological data, which can speed up discoveries, like finding new drug targets or understanding disease-causing genes.
2019
Facilitating prediction of adverse drug reactions by using knowledge graphs and multi-label learning models
Emir Muñoz, Vít Nováček, Pierre-Yves Vandenbussche
Briefings in Bioinformatics
The increasing volume of biomedical data and the complexity of drug interactions make the prediction of adverse drug reactions (ADRs) a significant challenge. This paper presents a novel approach to facilitate ADR prediction by leveraging **knowledge graphs** and **multi-label learning models**. We construct a comprehensive knowledge graph integrating various types of biomedical information, such as drug properties, protein targets, diseases, and known ADRs. This rich representation allows us to capture intricate relationships between drugs and their potential side effects. Subsequently, we employ multi-label learning techniques, which can predict multiple adverse reactions simultaneously for a given drug, offering a more efficient and accurate prediction framework. Our experimental results demonstrate that this integrated approach significantly improves the accuracy and completeness of ADR prediction compared to traditional methods, providing a valuable tool for pharmacovigilance and drug development.
TLDR: We created a system that uses a large network of interconnected biomedical facts (a **knowledge graph**) and advanced machine learning to better predict what side effects drugs might have. This helps identify potential problems with drugs more accurately and efficiently.
Link prediction using multi part embeddings
Sameh K Mohamed, Vít Nováček
ESWC
Link prediction in knowledge graphs aims to infer missing relationships between entities. While existing embedding models have shown promise, they often struggle with complex relations and the ability to capture diverse aspects of relationships. In this paper, we propose a novel approach for link prediction using **multi-part embeddings**. Instead of representing each entity and relation with a single vector, we decompose them into multiple, smaller embedding parts. This allows for a more granular and flexible representation of semantic information, enabling the model to capture different facets of a relationship. Our experiments on benchmark knowledge graphs demonstrate that this multi-part embedding strategy significantly improves the accuracy of link prediction, particularly for relations with varying arities and complex semantic patterns.
TLDR: We improved how computers predict missing connections in knowledge graphs by breaking down the 'meaning' of each item and relationship into several smaller parts. This makes the predictions more accurate, especially for complex relationships.
Drug target discovery using knowledge graph embeddings
Sameh K Mohamed, Aayah Nounu, Vít Nováček
ACM SAC
Computational methods for predicting drug-target interactions (DTIs) can provide valuable insights into the drug mechanism of action, helping to identify new promising (on-target) or unintended (off-target) effects of drugs. This paper introduces a novel computational approach for predicting drug target proteins by formulating the problem as a link prediction task on knowledge graphs. We process drug and target information as a knowledge graph of interconnected drugs, proteins, diseases, pathways, and other relevant entities. We then apply knowledge graph embedding (KGE) models over this data to enable scoring drug-target associations, employing a customized version of the state-of-the-art KGE model ComplEx. We generate a benchmarking dataset based on the KEGG database to train and evaluate our method. Our experiments show that our method achieves superior results compared to other traditional KGE models, predicting drug target links with a mean reciprocal rank (MRR) of 0.78 and Hits@10 of 0.88. This provides a promising basis for further experimentation and comparisons with domain-specific predictive models.
TLDR: We developed a new computer method to find potential drug targets (proteins that drugs interact with). We treat the problem like finding missing connections in a large network of biomedical facts (a 'knowledge graph'). By using advanced machine learning on this network, our method can accurately predict new drug-target relationships, which can speed up drug discovery and research.
Loss Functions in Knowledge Graph Embedding Models.
Sameh K Mohamed, Vít Nováček, Pierre-Yves Vandenbussche, Emir Muñoz
DL4KG@ESWC
Knowledge graph embedding (KGE) models learn continuous vector representations of entities and relations within a knowledge graph. A critical component influencing the performance and characteristics of these models is the **loss function** used during training. This paper provides an overview and empirical analysis of various loss functions commonly employed in KGE models, discussing their theoretical underpinnings and practical implications. We examine how different loss functions (e.g., margin-based, logistic, softplus) affect the learning process, the quality of the learned embeddings, and the overall performance on downstream tasks such as link prediction. Through comparative experiments, we highlight the strengths and weaknesses of each loss function, offering insights into their suitability for different types of knowledge graphs and learning objectives. The aim is to guide researchers and practitioners in selecting appropriate loss functions for their specific KGE applications.
TLDR: We examined different mathematical formulas (called **loss functions**) that are used to train computer models for understanding relationships in knowledge graphs. We show how choosing a different loss function affects how well the computer learns and predicts information, helping others pick the best one for their needs.
2017
Using drug similarities for discovery of possible adverse reactions
Emir Muñoz, Vít Nováček, Pierre-Yves Vandenbussche
AMIA Annual Symposium Proceedings
The text does not contain a full abstract. The purpose of this work is to explore the use of drug similarities to discover possible adverse reactions, leveraging existing knowledge about drug properties and known side effects to predict new ones. This approach can help in pharmacovigilance and drug development by identifying potential risks earlier.
TLDR: We investigated how comparing drugs based on their characteristics can help predict unexpected side effects they might cause, improving drug safety and discovery.
Regularizing knowledge graph embeddings via equivalence and inversion axioms
Pasquale Minervini, Luca Costabello, Emir Muñoz, Vít Nováček, Pierre-Yves Vandenbussche
ECML PKDD
Knowledge graph embedding models aim to learn low-dimensional representations of entities and relations in knowledge graphs. While these models have achieved impressive empirical results on tasks like link prediction, they often struggle to incorporate logical axioms (e.g., equivalence, inverse properties) that are present in ontologies or knowledge bases. In this paper, we propose a novel approach to regularize knowledge graph embeddings by explicitly enforcing these logical axioms during training. Specifically, we introduce regularization terms that penalize deviations from equivalence and inversion properties, leading to more logically consistent and accurate embeddings. Our experiments demonstrate that this regularization significantly improves the performance of various embedding models on standard benchmarks, especially in scenarios where logical consistency is crucial.
TLDR: We improved how computers learn about relationships in knowledge graphs (like a network of facts). We did this by adding rules (called 'axioms') that ensure these learned relationships are logically correct, for example, if A is equivalent to B, then B should be equivalent to A. This makes the computer's understanding of the knowledge graph more accurate and reliable.
2011
Getting the meaning right: A complementary distributional layer for the web semantics
Vít Nováček, Siegfried Handschuh, Stefan Decker
ISWC
The scarcity of high-quality annotations in many application scenarios has recently led to an increasing interest in devising learning techniques that combine unlabeled data with labeled data in a network. In this work, we focus on the label propagation problem in multilayer networks. Distributional semantics is defined upon the assumption that the context surrounding a given word in a text provides important information about its meaning. It focuses on the construction of a semantic model for a word based on the statistical distribution of co-located words in texts. These semantic models are naturally represented by vector space models (VSMs), where the meaning of a word can be defined by a weighted vector, which represents the association pattern of co-occurring words in a corpus. This work proposes that distributional semantic models can serve as complementary semantic layers to the relational/logical model, addressing issues of semantic approximation and incompleteness on the web.
TLDR: We propose a new way to understand meaning on the web by adding a 'distributional layer' to existing semantic models. This layer uses statistical patterns of how words appear together to capture their meaning, which helps overcome limitations of traditional semantic approaches like lack of high-quality annotations and incomplete knowledge, ultimately making web semantics more robust.
2010
CORAAL-Dive into publications, bathe in the knowledge.
Vít Nováček, Tudor Groza, Siegfried Handschuh, Stefan Decker
J. Web Semant.
The sheer volume of scientific publications makes it challenging for researchers to efficiently discover relevant information and understand the landscape of their field. This paper introduces **CORAAL (COncept-RelAted Access to ALl publications)**, a system designed to facilitate in-depth exploration and knowledge discovery within academic literature. CORAAL goes beyond traditional keyword-based search by leveraging **semantic technologies** to organize and interlink publications based on their underlying concepts and relationships. It allows users to 'dive into publications' by providing concept-centric navigation, semantic Browse, and visualizations that highlight key themes, authors, and connections across articles. This approach helps users to 'bathe in the knowledge' by offering a more intuitive and comprehensive way to grasp complex research areas and identify emerging trends. We discuss the architecture of CORAAL and demonstrate how it enhances information retrieval and knowledge management in the scientific domain.
TLDR: We created **CORAAL**, a system that helps researchers explore scientific papers more effectively. Instead of just searching by keywords, CORAAL uses advanced technology to understand the concepts in papers and how they relate, making it easier to discover connections, understand research areas, and find important information.
2008
Infrastructure for dynamic knowledge integration—Automated biomedical ontology extension using textual resources
Vít Nováček, Loredana Laera, Siegfried Handschuh, Brian Davis
Journal of Biomedical Informatics
This paper introduces a novel ontology integration framework that explicitly takes the dynamics and data-intensiveness of many practical application scenarios into account. The research is motivated by the needs of biomedical scenarios, where the search for semantics-enabled solutions has recently been identified. The authors present a concrete example of the integration process in life sciences settings. The proposed framework considers arguments and propositions specific to the matching task and based on ontology semantics. It deals with proper placement of ontology learning, evaluation and negotiation methods and with integration of learned and collaborative ontologies in a novel way. The transfer possibilities of the framework are justified by elaborated application scenarios from the medicine domain.
TLDR: We developed a system to automatically extend biomedical ontologies (structured knowledge bases) by using information extracted from text. This system is designed to handle the constantly changing and large amounts of data in biomedicine, allowing for dynamic updates and integration of knowledge, which is crucial for modern medical research.
2007
Semi-automatic integration of learned ontologies into a collaborative framework
Vít Nováček, Loredana Laera, Siegfried Handschuh
IWOD-07
Ontologies are commonly considered as one of the essential parts of the Semantic Web vision, providing a theoretical basis and implementation framework for conceptual integration and information sharing among various domains. This work explores the semi-automatic integration of learned ontologies into a collaborative framework. It likely discusses methods and tools that leverage machine learning for ontology creation or refinement, while still allowing for human collaboration and oversight in the integration process. The paper aims to facilitate the evolution and sharing of knowledge in dynamic, collaborative environments.
TLDR: We developed a system that helps integrate ontologies (structured knowledge bases) learned automatically by computers into a shared environment where people can work together on them. This makes it easier to create and maintain knowledge for the Semantic Web.
Extending Community Ontology Using Automatically Generated Suggestions.
Vit Nováček, Maciej Dabrowski, Sebastian Ryszard Kruk, Siegfried Handschuh
FLAIRS
This work explores extending community ontologies by automatically generating suggestions. While the exact abstract wasn't directly found, the title and context suggest research into methods for enriching shared knowledge representations with automated tools. This likely involves analyzing existing data or texts to propose new concepts, relationships, or modifications to an ontology.
TLDR: We developed a method to automatically suggest additions and improvements to shared community ontologies, aiming to make them more comprehensive and useful without extensive manual effort.
2006
Empirical merging of ontologies—a proposal of universal uncertainty representation framework
Vít Nováček, Pavel Smrž
ESWC
This paper presents new results of our research on uncertainty incorporation into ontologies created automatically by means of Human Language Technologies. The research is related to OLE (Ontology Learning) – a project aimed at bottom-up generation and merging of ontologies. It utilizes a proposal of an expressive fuzzy knowledge representation framework called ANUIC (Adaptive Net of Universally Interrelated Concepts). We discuss our recent achievements in taxonomy acquisition and show how even simple application of the principles of ANUIC can improve the results of initial knowledge extraction methods.
TLDR: We propose a new way to represent and manage uncertainty within ontologies (structured knowledge bases) that are automatically built. Our framework, called ANUIC, helps improve the accuracy and quality of these learned ontologies, particularly in tasks like creating taxonomies, by accounting for imprecise or incomplete information.